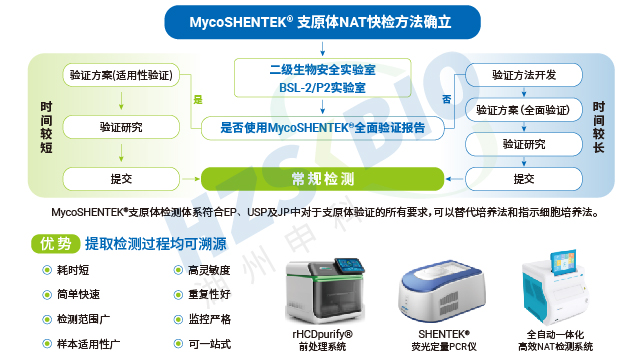
湖州申科 MycoSHENTEK® 支原体检测解决方案以合规性为中心,具备突出的验证优势。该方案的试剂盒已完成 FDA 的 DMF 备案,经过 FDA 严格审核确认准确性与合规性,企业可直接引用备案信息,简化海外申报流程,降低审查不确定性,减少材料准备周期。方案通过了与合规机构的三方验证,联合合规机构获取标准菌株盘、建立 GC 比检测方法,共同开展提取效率验证、引物设计优化、反应体系与仪器程序开发,并由合规机构指导参与室间验证及检测限、专属性、耐用性等关键性能验证,严谨性与专业性获得行业认可。此外,方案还能为企业提供 NDA/BLA 申报验证支持,降低申报风险,同时涵盖试剂盒、设备、菌株、检测验证服务、现场审计等全流程服务,一站式解决企业整合成本高、流程复杂的痛点。
支原体检测过程中,每批次培养基需做灵敏度测试,确保检测有效。吉林生物制品支原体检测国产替代
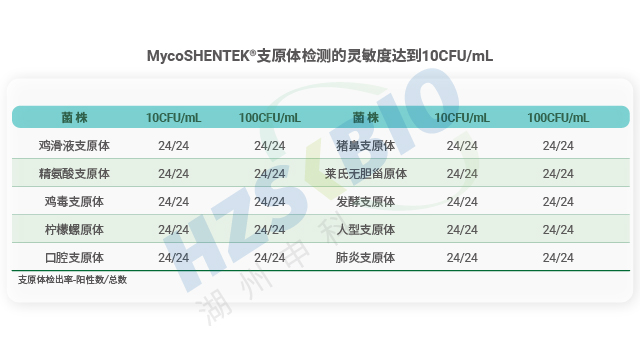
湖州申科的支原体验证菌株以高稳定性与合规性为重点优势,满足 NAT 方法验证需求。菌株来源可靠,均取自国内外合规机构验证菌株标准盘,溯源至美国 ATCC(口腔、肺炎支原体)、中国 CVCC(猪鼻支原体)等正规保藏机构,获正式商用授权,标定浓度涵盖 10CFU 与 100CFU。生产环境合规,在 BSL-2 生物安全实验室开展,符合国家生物安全法标准,针对不同菌株特性逐个优化生产工艺,涵盖超 10 种菌株的主代与工作代。质控环节严谨,采用高灵敏度培养基(液体、固体、半流体),保障菌落易观察分离与 CFU 计数准确性;冻存前后均通过固体平皿培养法测定 CFU,联合合规机构建立数字 PCR 标定方法,经实验室间对比验证,准确监控 GC/CFU 比,确保菌株质量达标。
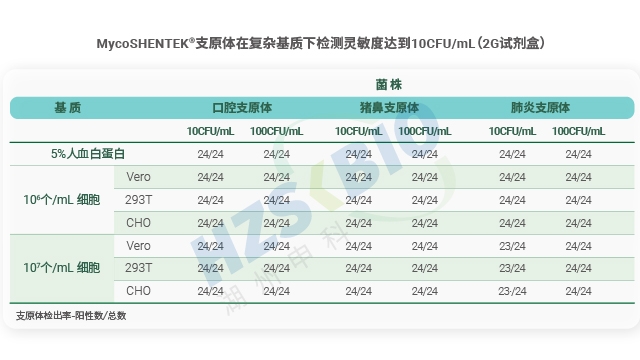
天津复杂基质支原体检测可比性验证支原体标准菌株对支原体检测试剂盒的选择、使用及验证至关重要。
细胞和基因治疗领域正加速发展,国内以 CAR-T、间充质干细胞、AAV 基因治疗等新型生物制品势头正盛。这类产品与传统制药差异明显,给支原体检测带来全新挑战:批产量小但批次多,多数待检测样品含高达 10⁷个活细胞,且基质复杂如高蛋白、全血、高浓度质粒等。更关键的是,新型生物制品终末灭菌难度极大,需从起始材料、原物料到全工艺过程严格控污,而支原体污染隐蔽性强、危害大,成为质量安全控制的主要痛点,也推动着检测方法向更高效、抗干扰的方向升级。
外源因子全自动核酸检测分析系统系统在数据追溯与合规管理方面进行了针对性设计,完美适配生物药行业的严格监管要求。系统内置三级权限管理机制,可对操作人员、检测项目、数据访问进行准确管控,同时具备完善的日志审计追踪功能,详细记录检测全流程的关键信息,确保数据可追溯、可核查,完全符合 21CFR Part11 法规要求。数据传输方面,系统支持与 LIMS 实验室信息管理系统无缝对接,实现检测数据的自动同步与集中管理,同时配备 USB 接口,可直接连接打印机打印实验结果,满足企业纸质记录存档需求。这些功能让支原体检测的每一个环节都处于合规管控之下,为企业应对监管检查、保障数据真实性提供了有力支持。
一体化支原体检测卡盒集成全流程,相当于迷你 qPCR 实验室,普通环境即可使用。
质控结果是支原体 NAT 检测可靠性的前提,需严格遵循判定标准,且需结合实验室实际条件验证适配的标准阈值。质控样品的判定规则明确:NTC(无模板对照)的 FAM 信号需 2 复孔 Ct≥40 或无明显扩增曲线,VIC 信号需 2 复孔 Ct<35 且呈有效 “S” 型扩增;NCS(阴性对照)判定标准与 NTC 一致;PC(阳性质控)的 FAM 信号需 2 复孔 Ct<35 且呈有效 “S” 型扩增,PCS(阳性对照底物)判定标准与 PC 一致。只有质控结果全部满足要求,才能进一步分析样本结果,若质控不达标,需排查设备、试剂、操作等环节的问题并重新检测。
支原体兼具胞内胞外生存特性,检测需裂解细胞而非只取上清,避免漏检胞内污染。湖北生物制品支原体检测技术服务
复杂样品在进行支原体检测时,可适当稀释样本或增加蛋白酶 K 用量,消除基质干扰。吉林生物制品支原体检测国产替代
抑制物质检测是支原体培养法的关键前置环节,旨在排除供试品中抑制成分对检测的干扰。湖州申科按 USP 标准执行:对供试品进行一次抑制物质检测,若生产方法发生变化可能影响支原体检测结果,需重复该检测。检测通过两组平行实验对比实现:一组培养基加入供试品,另一组不加供试品,同步开展营养特性测试,判断供试品是否含抑制物质。若检出抑制物质,需通过适当方法中和或抵消其作用,例如使用不含抑制剂的底物,或将供试品稀释在更大体积的培养基中,确保支原体能在检测体系中正常生长,避免因抑制成分导致检测结果失真。
吉林生物制品支原体检测国产替代