图2MAZTER-Seq实验流程图图3MAZTER-MINE分析m6A示意图接下来作者便是要验证这一新方法的可行性了。在酵母中敲除IME4的情况下,检测到的剪切效率高于野生型(剪切效率高低m6A水平),m6A抗体富集后的样品剪切效率也低于未富集的Input组。整体水平可靠,那检测的特异性位点是否准确呢?作者也将该方法检测到的新甲基化位点使用放射标记层析检测,发现预测的位点准确存在而且与剪切效率相符合。如图5所示。而图6中,作者则是与m6A抗体IP的方法进行了比较,也证实了这一方法的可行性。图5MAZTER-Seq检测结果验证图6MAZTER-Seq与m6A-Seq比较分析此外,后文中作者也在大规模的CRISPR-Cas9改变m6A状态和酵母减数分裂模型中检测了MAZTER-Seq这一系统;并进一步通过这一方法检测了哺乳动物不同细胞间m6A水平的保守性;也探究了去甲基化酶FTO对整体m6A甲基化水平的影响等。这里小编主要给大家分享这一新技术,其他部分暂不过多分析了。新的技术能拓展我们的研究内容;对于这一技术。纯干货Western blot (WB)条带灰度统计与GraphPad作图.山西科研技术服务

二甲苯Ⅰ、Ⅱ各15min至透明。(2)放入二甲苯和石蜡等量混合液处理15min,再放入石蜡Ⅰ、Ⅱ透蜡各50~60min。透蜡在恒温箱内进行,箱内温度保持在55~60℃左右。4、包埋(1)用镊子夹取蜡模在酒精灯上稍加热,并倒入少许从温箱中取出的纯石蜡。(2)再将镊子在酒精灯上稍加热,夹取材料将切面朝下放入蜡模中,排列整齐,再放上包埋盒,轻轻倒入熔蜡。5、切片、展片、贴片(1)将包埋好的石蜡块固定在切片机上,调整厚度调节器到所需的切片厚度,一般为4~6μm。(2)切下的组织薄片要放到加热的水中烫平,再贴到载玻片上,放45℃恒温箱中烘干。二、HE染色1、脱蜡、复水(1)保持水浴锅温度为60℃,将切片放入干燥的染色缸内,放入水浴锅中,30min至蜡熔化。(2)石蜡切片经二甲苯Ⅰ、Ⅱ脱蜡各5min,然后放入100%、95%、90%、80%、70%各级酒精溶液中各3~5min,再放入蒸馏水中3min,以便染料可以进入组织。2、染色、脱水(1)切片放入苏木精中染色约10~30min,用流水冲洗约15min,使切片颜色变蓝。(2)将切片放入1%盐酸乙醇液中褪色,几秒后见切片变红、颜色较浅即可,后将切片再放入流水中使其恢复蓝色。(3)切片放入50%、70%、80%乙醇中各3~5min。北京外包科研技术服务购买细胞的结构是细胞生物学的重要研究内容之一。
准备碎冰和。把膜置于甲醇溶液中活化1分钟,然后转移至转膜液中平衡3分钟后备用。2、转膜组装转膜三明治夹,这一步很关键,重要的是胶和膜之间不能有气泡且膜与胶要保证贴合紧密(否则会翻车),可以加多点转膜液泡着。组装好后加满转膜液,设置电源参数,我习惯恒流转膜,200-280毫安,1-2小时,根据分子量定,如50KD的分子用60分钟就够了。如果一个电源带两个转膜大槽即四块胶,我就会用恒压70-110V。这个过程重要的是做好降温。这里简单说一下蛋白分子量与玻璃板厚度,分离胶的浓度,转膜电源参数的选择问题。如果是能用1毫米的玻璃板就不用,因为转膜是在电场的作用下蛋白分子从胶上迁移到膜上,1毫米胶的蛋白迁移距离要比。选择更薄的胶蛋白转膜时间可以减少,从而减少发热,以免胶变形,条带也会更好看。然后是分离胶的浓度,如果是150—200KD的分子选10%以下的的分离胶,200—300KD的选8%的,300KD以上的选6%的,小于30KD的200mA30分钟,30-100KD的按分子量的数值算,如70KD,250mA70分钟;100-150KD的250mA100分钟;150—300KD的分子转膜条件用280毫安(以上均是对于,),120分钟足矣,前提是胶的浓度相适应。五、封闭孵一抗1、封闭转膜结束后。
静置25分钟后把酒精倒干,用吸水纸吸出多余的酒精,然后配压缩胶,同样的操作,关键是梳子要插得快,要小心梳子下产生气泡,然后静置30分钟。如果是当天跑胶,我会等上层胶凝2个小时再用,但要注意防干燥缩水,可以在一个小时的时候沿着梳子上缘加点电泳液。所以我一般提前一晚制胶,泡于纯水或者电泳液里置于4度冰箱暂存。三、蛋白电泳1、上样前准备把胶组装到电泳芯上,注意密闭性(否则漏液),如果内槽漏液就不是匀强电场了,条带可能就不是一条直线。然后内槽倒满电泳液,拔梳子,这一步要小心,梳子要两边一起缓缓往上拔出,然后观察泳道内有无脱落的胶粒或者胶丝,有的话用1毫升注射器吸出。然后从冰箱取出蛋白样品,解冻。准备振荡器。2、上样和电泳注意,上样后蛋白会开始慢慢在胶中弥散,所以上样越快越好。我习惯先上蛋白Marker,再上蛋白样品,蛋白上样前确保样品完全解冻和充分振荡(推荐使用振荡器振荡),吸的时候没有拉丝即可,建议上样分钟把样品从冰上取出来,不然样品中SDS可能会结晶析出,从而影响电泳效果。上层胶80V25分钟,下层胶120V65分钟。四、转膜1、转膜前准备我会在电泳结束0分钟准备,把转膜液配好置于4度冰箱预冷,然后裁膜,准备转膜装置。细胞是生命的基本单位,所有生物体都是由一个或多个细胞组成的。下面就跟着上海东寰一起看看吧。
应退回纯酒精中重新脱水,然后再透明,否则切片难以镜检。(4)二甲苯应尽量保持无水,应经常更换,或用纱布包无水硫酸铜放入染色缸内吸收水分。5.透蜡注意事项(1)透蜡的目的是除去组织中的透明剂(如二甲苯等),使石蜡渗透到组织内部达到饱和程度以便包埋。透蜡时间根据组织大小而定。(2)尽量保持在较低温度中进行,以石蜡不凝固为度。(3)透蜡温度要恒定,不可忽高忽低。(4)操作要迅速,力求在短的时间内完成石蜡透入过程,以免引起组织变硬、变脆、收缩等。(5)注意温度不要过高,以免组织发脆。6.染色水洗注意事项(1)染色原理:伊红主要染细胞质,着色浓淡应与苏木精染细胞核的浓淡相配合,如果细胞核染色较浓,细胞质也应浓染,以获得鲜明的对比。(2)染色时间应根据染色剂的成熟程度及室温高低,适当缩短或延长。室温高时促进染色,染色时间可短些,否则可适当延长时间,冬季室温低时可放入恒温箱中染色。(3)注意流水不能过大,以防切片脱落。(4)如果细胞核染色较浅,细胞质也应淡染。可在伊红乙醇液中滴加数滴冰醋酸助染,促使细胞质容易着色,并且经乙醇脱水时不易褪色。此外,动物模型的伦理问题也不容忽视,科研人员需要在符合伦理规定的前提下进行相关研究。海南大鼠科研技术服务购买
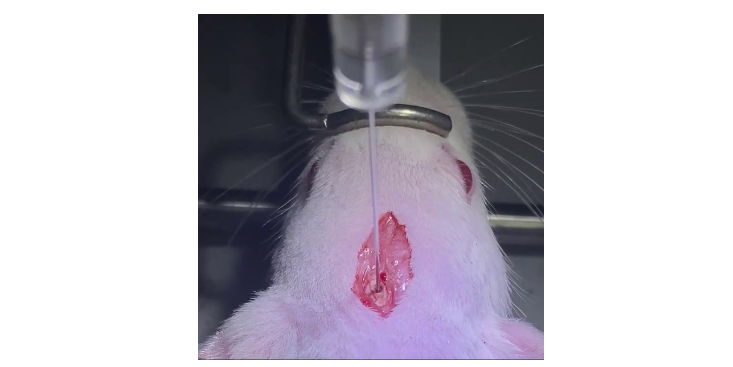
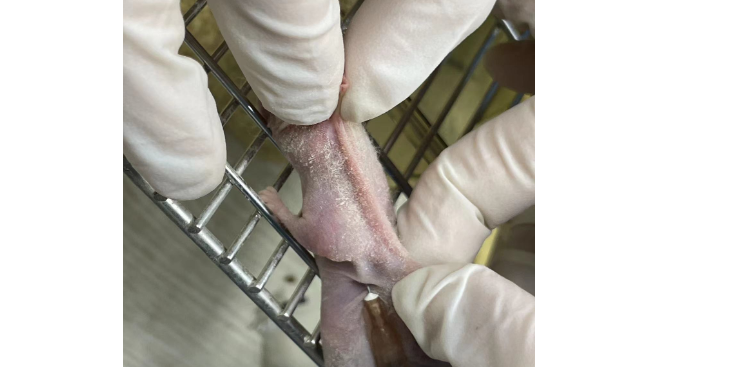
肿瘤细胞具有极强的增殖能力,在一个适宜,裸鼠成瘤是常见的验证肿瘤细胞体内增殖模型。山西科研技术服务
建议按照2×106每孔的数量将293T细胞均匀铺入。(2)第二天:在24小时之内,观察293T细胞的汇合度在90%~95%之间时,向其中加入DNA-脂质体复合体,DNA-脂质体复合体制备方法如下:a)轻轻混匀LipoMax,根据说明书加入相应量于500µlOpti-MEM无血清培养基中,混合均匀并置于室温5分钟。b)在500µlOpti-MEM无血清培养基中稀释DNA,总质量为15µg按照载体质粒:psPAX2:=4:3:1的比例加入DNA。c)将稀释后的LipoMax和稀释后的DNA轻轻混匀,常温静置20分钟,形成DNA-LipoMax复合体。(3)将DNA-LipoMax复合体轻柔地滴加至细胞培养皿中,轻轻摇晃培养皿混匀,放入细胞培养箱中培养。(4)病毒收集浓缩病毒:加入DNA-LipoMax复合体48小时后,收集病毒上清,同时加入10ml预温的293T培养基到细胞培养皿中。将收集到的病毒上清存在4℃冰箱中;收集72小时病毒上清,与48小时病毒上清混在一起。将离心机温度降温到4℃,600g,离心5分钟,去除其中的细胞碎片,上清液经µm滤头过滤,加入病毒浓缩液,配制浓缩病毒液。将浓缩后的病毒放于4℃冰箱摇床上,旋转过夜。第二天,4度离心机,3000~4000g离心15分钟。弃掉上清液,加入1Xpbs或培养基重悬。山西科研技术服务